Computational Comparison of the Binding Affinity of Selective and Nonselective NSAIDs to COX-2 Using Molecular Docking

Doménica Flores 1 , Carola Jerves1
1 Universidad de Cuenca/ Departamento de Química Aplicada/ Cuenca/ Ecuador;
; domenicaa.flores@ucuenca.edu.ec
; domenicaa.flores@ucuenca.edu.ec
*Correspondence: carola.jerves@ucuenca.edu.ec
ABSTRACT
Cyclooxygenase-2 (COX-2) plays a key role in inflammation, making it a prime target for nonsteroidal anti-inflammatory drugs (NSAIDs). This study uses molecular docking to compare the binding affinities of four nonselective NSAIDs (aspirin, ibuprofen, diclofenac, naproxen) and three selective COX-2 inhibitors (celecoxib, rofecoxib, etoricoxib) to COX-2. Simulations with AutoDock4 and AutoDock Vina revealed distinct differences in binding profiles and selectivity. Selective COX-2 inhibitors exhibited stronger binding affinities, with etoricoxib achieving -11.22 kcal/mol (AutoDock4), driven by key hydrogen bonds and π interactions. Nonselective NSAIDs, such as diclofenac (-8.08 kcal/mol), showed moderate affinity but lacked specificity, targeting both COX isoforms and increasing gastrointestinal side effects. AutoDock4 provided detailed conformational analysis, while AutoDock Vina complemented with faster but less detailed results. This research highlights the structural interactions underlying NSAID efficacy and side effects, offering valuable insights for drug design. Selective inhibitors provide improved safety profiles for long-term use, while nonselective NSAIDs remain effective for short-term treatments. These findings emphasize the importance of computational tools in optimizing NSAID selectivity and efficacy, paving the way for developing safer anti-inflammatory therapies.
Keywords: NSAIDs, active site, COX-2, Docking, selectivity.
Keywords: NSAIDs, active site, COX-2, Docking, selectivity.
INTRODUCTION
Cyclooxygenase (COX) enzymes are pivotal in the biosynthesis of prostaglandins, which regulate inflammation, pain, and fever1. The two primary isoforms, COX-1 and COX-2, differ significantly in their expression and physiological roles. COX-1 is constitutively expressed in most tissues, where it maintains homeostatic functions, such as protecting the gastrointestinal lining and ensuring renal blood flow2. Conversely, COX-2 is inducible, expressed primarily during inflammation, making it a key target for anti-inflammatory therapies to reduce pain and swelling with minimal adverse effects on normal physiological processes1,3.
A significant difference between the active sites of COX-1 and COX-2 lies in their size and structural composition. The active site of COX-1 features an isoleucine residue at position (Ile434), which limits the accessibility of more significant inhibitors. In contrast, COX-2 has a valine residue at the corresponding position (Val523), creating a larger, more flexible binding pocket (Figure 1). This structural variation allows selective COX-2 inhibitors, or coxibs, to specifically bind to COX-2 due to the larger size of its active site, enhancing their selectivity and reducing gastrointestinal and renal side effects. Traditional NSAIDs, being smaller, can bind to both COX-1 and COX-2 active sites, compromising their selectivity and leading to increased adverse effects associated with the inhibition of COX-14.
A major difference between the active sites of COX-1 and COX-2 lies in their structural composition. The active site of COX-1 contains an isoleucine residue at position 434, which restricts access to bulkier inhibitors. In contrast, COX-2 has a valine residue at the corresponding position (Val523), resulting in a larger, more flexible side pocket4 (Figure 1). This structural distinction allows selective COX-2 inhibitors, or coxibs, to bind specifically to COX-2 while sparing COX-1. By selectively targeting COX-2, these drugs reduce the gastrointestinal and renal side effects commonly associated with nonselective NSAIDs. While coxibs agents are safer for the gastrointestinal system, they may increase cardiovascular risks, highlighting the need for balanced therapeutic approaches5–7.
Molecular docking is a computational tool that enables detailed simulation of the interactions between drugs and their biological targets. This method provides insights into the binding mechanisms of NSAIDs within the active sites of COX enzymes, evaluating parameters such as binding free energy and inhibition constants. These metrics are crucial for understanding drug selectivity and efficacy. This study employs molecular docking to compare the binding affinities of selective and nonselective NSAIDs to COX-2, aiming to elucidate the molecular basis of their therapeutic actions and potential side effects8–11.
The structural insights gained through docking studies have been instrumental in refining the design of selective COX-2 inhibitors, optimizing their efficacy while minimizing adverse effects. By examining the molecular interactions of NSAIDs with COX-2, this research contributes to developing safer anti-inflammatory agents. Such advancements hold promise for creating therapies tailored to specific patient needs, addressing the limitations of current treatments, and improving outcomes for inflammatory conditions12.
This study underscores the value of computational pharmacology in drug development. By analyzing the differential binding behaviors of NSAIDs, it lays the groundwork for the rational design of next-generation anti-inflammatory drugs. The findings deepen our understanding of the molecular interactions that govern NSAID selectivity and support the optimization of therapeutic profiles to achieve a more favorable balance between efficacy and safety. Additionally, this study provides a basis for comparing in silico results with clinical findings, helping to bridge the gap between computational predictions and real-world therapeutic outcomes.
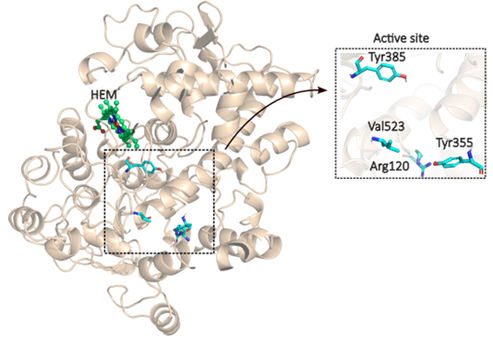
Figure 1. The active site of the enzyme COX-2.
MATERIAL AND METHODS
The selection of NSAIDs for this study was based on previous local research identifying the most commonly used anti-inflammatory drugs in both self-medication and chronic pain management. Studies by Jara et al.13 and Encalada et al. 14 provided valuable insight into NSAID consumption patterns in Ecuador. Jara et al. reported that 69.6% of households in the San Blas parish had self-medicated with NSAIDs, while Encalada et al. found that 26% of older adults across 15 urban parishes in Cuenca engaged in NSAID self-medication. Given their frequent use, particularly for pain management, seven NSAIDs were selected for in silico analysis: four conventional, nonselective COX inhibitors (acetylsalicylic acid, ibuprofen, diclofenac, and naproxen) and three selective COX-2 inhibitors (celecoxib, rofecoxib, and etoricoxib). The three-dimensional structures of the selected compounds were obtained from the PubChem database in .sdf format. Each ligand underwent geometric optimization and energy minimization using the MMFF94 force field in Avogadro software (version 1.2.0)15, ensuring their conformations were energetically favorable for docking simulations.
The COX-2 protein structure (PDB ID: 1CX2) was retrieved from the Protein Data Bank. This structure, which includes the enzyme's heme cofactor, was complexed initially with the inhibitor SC-558. To prepare the protein for docking simulations, the SC-558 molecule was removed, along with unnecessary water molecules and other non-essential elements. Hydrogen atoms were added to stabilize the molecular structure and enable more accurate predictions of ligand binding interactions. The processed protein structure was saved in .pdbqt format, suitable for docking simulations. It is important to note that this study did not involve a comparative analysis of different crystallographic structures COX-2, as the focus was solely on evaluating the binding affinity and interaction profiles of NSAIDs using a single, well-characterized protein model.
Docking experiments were conducted using AutoDock Tools (version 1.5.7), which integrates both AutoDock416 and AutoDock Vina17. This combination was chosen to take advantage of the strengths of each platform: AutoDock4's Lamarckian genetic algorithm, known for its robust conformational sampling, and AutoDock Vina's gradient-based algorithm, which offers enhanced computational efficiency and speed. The Lamarckian genetic algorithm was selected due to its adaptive learning capabilities, allowing ligand conformations to evolve towards the most favorable binding poses over successive iterations and improving docking accuracy. The COX-2 enzyme and the ligands were treated as flexible to capture the dynamic nature of ligand-protein interactions, particularly in the key active-site residues. This flexibility allowed the NSAIDs to explore various conformations within the active site, yielding a more physiologically relevant binding profile. Each ligand was docked separately into the COX-2 active site, with AutoDock4 utilizing the Lamarckian genetic algorithm, optimized for conformational sampling through parameters like population size, mutation rate, and energy evaluations. AutoDock Vina was employed for repeated docking experiments to provide comparative results. To ensure the lowest energy states were identified, each ligand was docked twice in each software, guaranteeing that the most favorable binding energies were captured. Both software tools provided binding free energy and inhibition constant values, which were analyzed to assess the binding affinities of the NSAIDs.
The docking grid was configured to include key residues in the COX-2 active site, identified from previous structural studies as Arg120, Tyr355, Tyr385, and Val523. The grid dimensions were set at 50x50x50 Å with a grid spacing of 0.414 Å, ensuring comprehensive binding site coverage. The grid box was centered at x = 24.447, y = 22.535, and z = 16.327 to capture all potential binding interactions between the ligands and the enzyme's active site residues.
Post-docking analysis involved multiple tools to interpret and visualize the results. Binding energies and inhibition constants were statistically analyzed in RStudio (version 1.4.1106)18 to compare the performance of selective and nonselective NSAIDs. Visualization of interactions between the ligands and COX-2 was conducted in PyMOL (version 3.0.3)19 for three-dimensional representations, while Discovery Studio 202120 was used to generate detailed two-dimensional diagrams of the interactions, categorizing them as hydrogen bonds, hydrophobic interactions, or π-stacking.
RESULTS
Molecular docking analyses revealed distinct differences in the binding affinities and interaction profiles of selective and nonselective NSAIDs with COX-2. Selective NSAIDs, such as etoricoxib, exhibited consistently stronger interactions, with an average binding free energy of -10.78 ± 0.50 kcal/mol in AutoDock4. These compounds effectively engaged the Val523 residue, allowing access to the enzyme's side pocket and stabilizing the ligand-enzyme complex. This selectivity minimizes off-target interactions with COX-1, reducing adverse effects.
In contrast, nonselective NSAIDs displayed lower binding affinities, averaging -6.69 ± 1.51 kcal/mol in AutoDock4. Although they interacted with the COX-2 active site, their inability to target Val523 specifically resulted in broader inhibition of both COX isoforms. This dual inhibition contributes to the increased gastrointestinal and renal side effects associated with nonselective NSAIDs. However, diclofenac's relatively strong interaction with COX-2 supports its efficacy as an anti-inflammatory agent, albeit with a less favorable safety profile. The results of each drug are reflected in Table 1.
Overall, the results emphasize the therapeutic trade-offs between efficacy and selectivity. While selective inhibitors offer enhanced safety for long-term use, nonselective NSAIDs remain potent options for short-term anti-inflammatory therapy when gastrointestinal risks are managed.
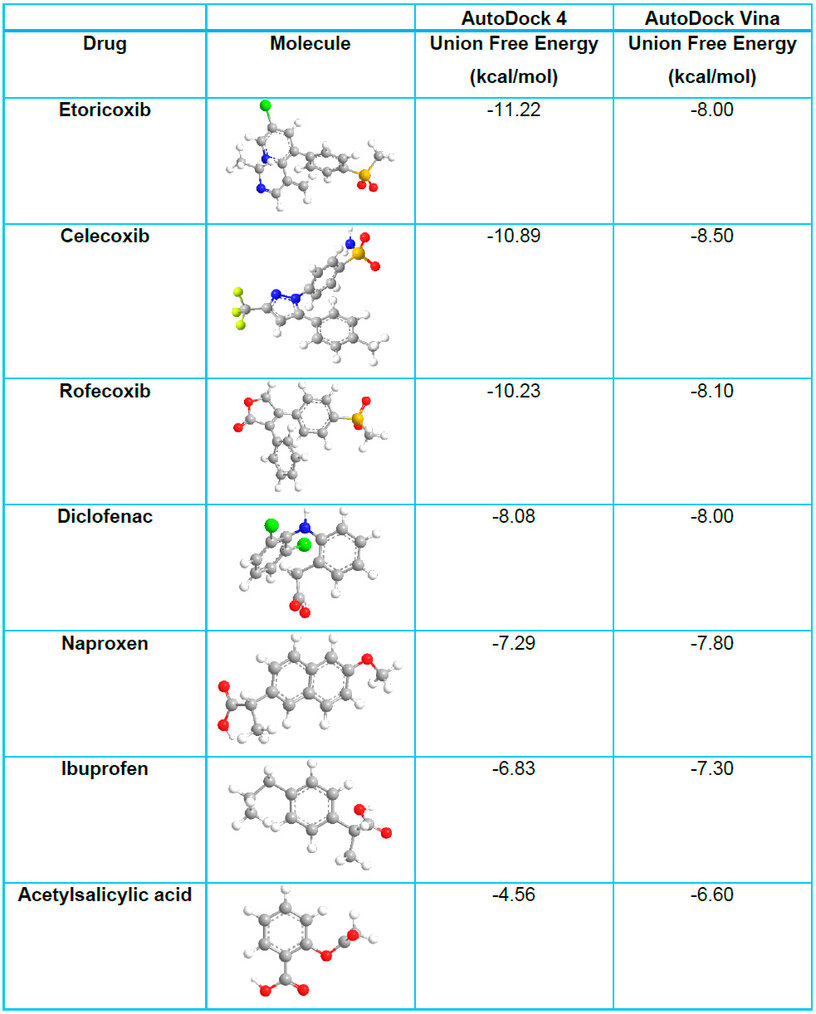
Table 1. AutoDock 4 vs. Autodock Vina Union Free Energy Values.
Statistical analysis further confirmed the observed differences. The mean binding free energy and inhibition constant for each group of NSAIDs obtained in AutoDock 4 were calculated, excluding aspirin from the nonselective group due to its distinct mechanism of action. A t-test performed in RStudio yielded a p-value of 0.008, indicating a significant difference in binding affinities between selective and nonselective NSAIDs; the averages obtained are shown in Table 2. This difference reflects their pharmacological behavior, as selective inhibitors demonstrate a greater affinity for COX-2, leading to a more targeted inhibition of prostaglandin synthesis while preserving COX-1 activity, thereby reducing gastrointestinal and renal risks. In contrast, nonselective NSAIDs, by inhibiting both isoforms indiscriminately, are associated with higher toxicity, particularly in long-term use.
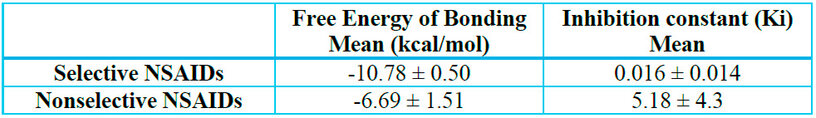
Table 2. Comparison of mean binding free energy values for COX-2 selective and nonselective NSAIDs.
Additionally, a comparison of mean binding free energy values between the two docking software programs was performed for both drug classes, shown in Table 3. A t-test for selective NSAIDs produced a p-value of 0.005, suggesting significant differences between the two software outputs. This discrepancy may be attributed to the increased structural complexity of selective inhibitors, which could lead to variations in results depending on the algorithm used. Conversely, a p-value of 0.9815 indicated no significant differences between the software-generated values for nonselective NSAIDs, suggesting a more consistent binding prediction for these compounds.
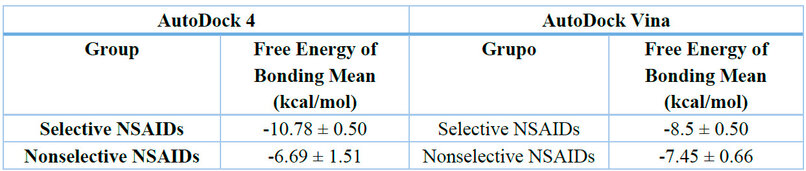
Table 3. Average Free Binding Energy of Selective and Nonselective NSAIDs according to AutoDock4 and AutoDock Vina.
These findings highlight key physiological and therapeutic implications. The significantly stronger binding of selective NSAIDs suggests their capacity to interact with COX-2 more precisely, leading to effective inflammation and pain reduction while minimizing COX-1 inhibition. This selectivity translates into a more targeted therapeutic effect with improved safety. Meanwhile, nonselective NSAIDs, despite their broader inhibition profile, provide adequate symptom relief, making them viable options for short-term treatment when gastrointestinal risks are managed appropriately. These results underscore the importance of molecular interactions in drug efficacy and reinforce the necessity of optimizing selectivity in NSAID design to balance therapeutic benefit and safety.
Diclofenac
Diclofenac, a nonselective NSAID, exhibited notable stability in its interaction with COX-2, with a binding free energy of -8.08 kcal/mol in AutoDock4 and -7.80 kcal/mol in AutoDock Vina. Docking analysis revealed several significant interactions within the active site of the enzyme. These interactions can be observed in 2D in Figure 2 and in Figure S1 of the supplementary material, which shows the interactions in 3D. Three hydrogen bonds were identified as pivotal for the stability of the ligand-enzyme complex. A non-conventional hydrogen bond was observed between the side chain of Arg120 and the hydroxyl oxygen of diclofenac at a distance of 2.0 Å. Another bond formed between the hydrogen of the aniline group in diclofenac and the carboxyl oxygen of Val523 at 2.5 Å. Additionally, the hydroxyl hydrogen of Tyr355 interacted with the carbonyl oxygen of diclofenac at a distance of 2.0 Å.
Visualization in three-dimensional space further demonstrated the complexity of interactions, highlighting a combination of covalent and non-covalent forces stabilizing the ligand. π-interactions played a significant role in enhancing binding. A π-σ interaction occurred between the benzene ring of diclofenac and the methyl side chain of Val523. Moreover, an amide-π stacked interaction was detected between the amide group of Gly526 and the second aromatic ring of diclofenac.
Hydrophobic interactions were also present, contributing to the overall binding energy. Alkyl-π interactions involved residues Phe518, Leu352, and Ala527, with Phe518 interacting via its benzene ring and diclofenac's alkyl group, while the benzene ring of diclofenac interacted with the methyl side chains of Leu352 and Ala527. Alkyl-alkyl interactions were observed with residues Val349 and Met522, where diclofenac's alkyl groups interacted with the side chains of these amino acids.
Finally, Van der Waals forces provided additional stabilization between diclofenac and multiple residues, including Val116, Tyr348, Ser353, Phe381, Tyr385, Trp387, Ser530, and Leu531. These interactions collectively highlight the multi-faceted binding profile of diclofenac, reflecting its potency and broad binding capacity within the COX-2 active site while explaining its efficacy and associated off-target effects.
The fluctuation in binding energy across different conformations of diclofenac was relatively low, suggesting consistent interactions with COX-2 across multiple binding poses. This stability aligns with its clinical use as a reliable anti-inflammatory and analgesic agent. However, its lack of COX-2 selectivity underscores the risk of adverse effects, particularly in long-term or high-dose therapies
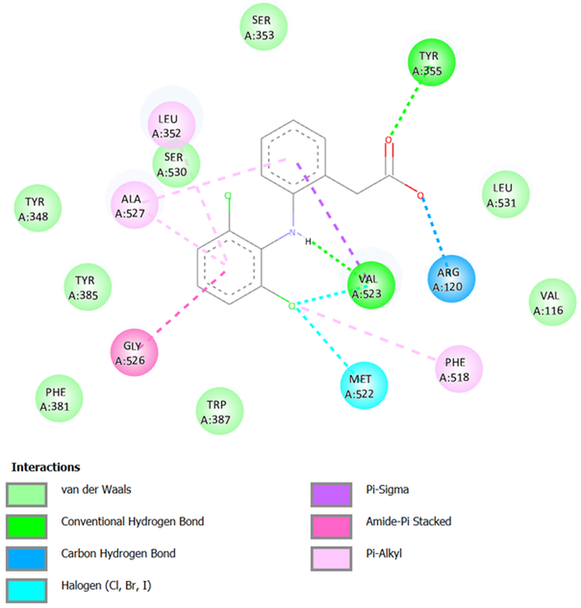
Figure 2. Interactions between Diclofenac and COX-2.
Etoricoxib
Etoricoxib, a selective COX-2 inhibitor, exhibited a strong binding affinity towards the enzyme, with binding free energies of -11.22 kcal/mol in AutoDock4 and -8.00 kcal/mol in AutoDock Vina. The interactions can be observed in 2D in Figure 3 and Figure S8 of the supplementary material, which shows the interactions in 3D. Docking analysis revealed two crucial hydrogen bonds that contribute significantly to the stability of the ligand-enzyme complex. The first hydrogen bond was formed between the hydrogen attached to the imidazole nitrogen of His90 and one oxygen atom of the methylsulfonyl etoricoxib group, with a distance of 1.9 Å. The second hydrogen bond involved the imine hydrogen of Arg513 and the second oxygen atom of the methylsulfonyl group at a distance of 2.2 Å.
In addition to hydrogen bonding, etoricoxib engaged in a variety of Van der Waals interactions, which were observed with residues Val116, Arg120, Gln192, Tyr355, Ala516, Gly525, and Ser530. Though weaker than hydrogen bonds, these interactions provided essential stabilization by ensuring the proper positioning of the ligand within the COX-2 active site.
Etoricoxib also exhibited several types of π interactions, further enhancing its binding specificity and stability. π-σ interactions were observed between the σ bonds of Ser353 and Val523 and the benzene ring of etoricoxib, as well as between Ala527 and Val349 and the pyridine ring of the molecule. A π-sulfur interaction was formed between the imidazole ring of His90 and the sulfur atom of the methylsulfonyl group in etoricoxib. Furthermore, a π-π stacked interaction occurred between the aromatic ring of Phe518 and the second pyridine ring of the ligand.
Additional interactions involved alkyl and π-alkyl interactions. These were observed between the aromatic rings of Phe518 and Trp387 and the alkyl group attached to the pyridine ring of etoricoxib. Similarly, alkyl groups of Leu352 (isobutyl), Val523 (isopropyl), and Ala527 (methyl) interacted with the methyl group of the non-chlorinated pyridine ring. Lastly, non-covalent alkyl interactions were formed between the methyl side chain of Met522 and the methyl group of the ligand.
The combination of hydrogen bonds, Van der Waals forces, and π interactions underpin the high specificity of etoricoxib for COX-2, emphasizing its role as a potent inhibitor. These interactions also explain its reduced off-target activity on COX-1, contributing to its improved gastrointestinal safety profile while maintaining a robust anti-inflammatory effect.
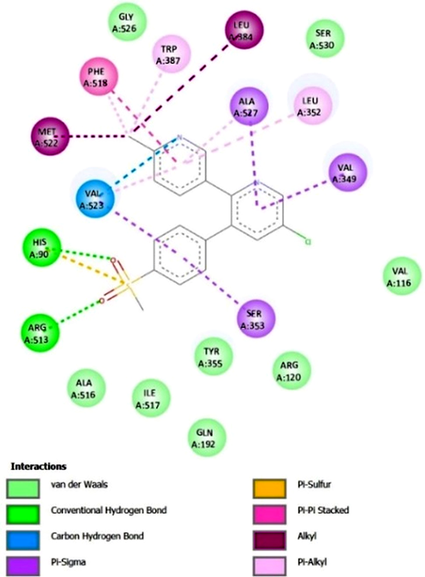
Figure 3. Interactions between Etoricoxib and COX-2.
The interactions of the other analyzed drugs can be found in the supporting material, Figures S1-S12.
DISCUSSION
The results of this study highlight the fundamental differences between selective and nonselective NSAIDs in their interactions with COX-2. Nonselective NSAIDs, such as diclofenac, exhibit moderate binding affinities and interact with both COX isoforms, resulting in practical anti-inflammatory effects but with increased gastrointestinal and renal risks due to COX-1 inhibition. In contrast, selective NSAIDs like etoricoxib demonstrate higher binding affinities and specificity for COX-2, minimizing adverse effects on COX-1-regulated physiological processes though potentially increasing cardiovascular risks. These findings underscore the importance of understanding molecular interactions to guide the clinical use of these drugs.
Several studies have reported similar results when comparing these findings with the literature. For instance, Bello-Vargas et al.21 analyzed ibuprofen, celecoxib, and diclofenac and found that the binding interactions for ibuprofen showed similarities with those reported in this study, particularly in the involvement of amino acids such as Val349 and Ala527. The study also reported that celecoxib demonstrated a higher affinity for COX-2, consistent with the findings of this study, where selective inhibitors exhibited stronger binding to COX-2 compared to nonselective NSAIDs.
In a study by Chaudhary & Aparoy22, molecular dynamics simulations were used to evaluate the binding energies of various selective COX-2 inhibitors. Their findings on celecoxib's binding interactions were similar to the results of this study, especially regarding the involvement of key amino acids such as Leu352, Arg513, and Phe518. However, this study also found additional interactions not previously reported, such as hydrogen bonds with Gln192, His90, Arg120, and Tyr355, suggesting that the binding profile of celecoxib may be more complex than initially understood.
Further comparison with Ibrahim et al.23 revealed similar binding energy values for diclofenac but differences in the binding affinity of rofecoxib. While Ibrahim et al. reported a binding energy of -8.241 kcal/mol for rofecoxib, this study found a stronger interaction between rofecoxib and COX-2, with values of -10.23 kcal/mol (AutoDock 4) and -8.50 kcal/mol (AutoDock Vina). These discrepancies highlight the variability in docking results across different studies and emphasize the importance of using multiple docking tools to obtain a more comprehensive understanding of drug-enzyme interactions.
Meneses & Cuesta24 provided another valid comparison, reporting binding energies for various NSAIDs that were similar to the results of this study. For instance, the binding energies of celecoxib, etoricoxib, ibuprofen, naproxen, and diclofenac were comparable across their research and this one, with slight variations attributed to the preparation of ligands and docking conditions. This consistency further supports the relevance and reliability of the results obtained in this study.
In summary, while there are some differences when comparing the findings of this study with those of previous research, the overall results align well with the existing literature. These findings suggest that the molecular docking techniques used in this study provide valuable insights into the binding affinities of selective and nonselective NSAIDs toward COX-2. Furthermore, the study underscores the importance of selectivity in NSAID design, as selective COX-2 inhibitors are known to reduce the gastrointestinal and renal side effects commonly associated with nonselective NSAIDs. The observed binding affinity values highlight the potential for developing safer and more effective anti-inflammatory drugs by optimizing their selectivity towards COX-2, which may ultimately lead to better patient therapeutic outcomes.
Diclofenac demonstrated a binding free energy of -8.08 kcal/mol in AutoDock4 and -7.80 kcal/mol in AutoDock Vina, reflecting its stable interaction with COX-2. Its ability to form three hydrogen bonds with Arg120, Val523, and Tyr355, alongside π interactions and hydrophobic contacts, supports its effectiveness as a nonselective NSAID. However, the lack of COX-2 specificity observed in its binding profile explains the associated risk of adverse effects. On the other hand, etoricoxib exhibited superior binding affinity with -11.22 kcal/mol in AutoDock4 and -8.00 kcal/mol in AutoDock Vina. Its selective binding to COX-2, facilitated by hydrogen bonds with His90 and Arg513 and enhanced by π interactions with Val523, Phe518, and other residues, underscores its role as a highly selective COX-2 inhibitor.
Differences in binding free energies were evident when comparing the results obtained from AutoDock4 and AutoDock Vina. AutoDock4 consistently produced lower energy values, likely due to its genetic algorithm-based sampling, which provides a more detailed exploration of conformational space. In contrast, AutoDock Vina's gradient-based approach offers faster processing but may lack the precision needed for exhaustive interaction analysis. Consequently, AutoDock4 was used as the primary tool for studying ligand-enzyme interactions in this research. While AutoDock Vina provided complementary energy values, its output lacked the detailed interaction data required for visualizing and understanding binding mechanisms, limiting its utility for in-depth molecular analysis.
This study employed flexible docking to account for ligand flexibility while maintaining the receptor in a rigid conformation. This approach was particularly relevant for analyzing the interaction of NSAIDs with COX-2, as these compounds exhibit conformational adaptability that can influence binding affinity and selectivity25. By allowing the ligand to adopt different conformations within the active site, flexible docking provides a more comprehensive understanding of molecular interactions than rigid docking, which may overlook relevant binding poses.
However, flexible docking methodologies present inherent limitations that must be considered when interpreting results. One major challenge is the inability to capture protein conformational changes upon ligand binding entirely. Although COX-2 undergoes structural adaptations during inhibitor binding, most docking algorithms, including those used in this study, treat the protein as a static entity. This limitation can lead to inaccuracies in predicting the proper binding mode, particularly for compounds that induce significant conformational rearrangements in the receptor25.
Another critical issue is the accuracy of scoring functions. While docking algorithms estimate binding affinity based on molecular interactions, current scoring functions may not always distinguish between near-native and incorrect binding poses with high precision. This limitation is particularly relevant for NSAIDs, where slight differences in binding orientation can significantly impact selectivity for COX-1 or COX-2. Moreover, while flexible docking allows ligands to explore different conformations, the receptor remains rigid, which may not fully capture induced-fit effects contributing to binding specificity25,26.
Computational complexity also represents a limitation, as flexible docking requires extensive conformational sampling to explore ligand flexibility, increasing processing time and computational demand. This constraint is particularly relevant when screening large libraries of compounds or performing exhaustive binding analyses. Additionally, the available structural data for COX-2 in complex with NSAIDs, while extensive, may still not fully represent all biologically relevant conformations, further impacting docking accuracy26.
CONCLUSIONS
Despite these limitations, flexible docking remains a valuable tool for understanding NSAID-COX-2 interactions, providing insights into binding affinity, selectivity, and potential structural modifications for drug optimization. The findings of this study align with previous reports, supporting the validity of computational docking approaches in analyzing NSAID binding properties. However, integrating docking with complementary techniques such as molecular dynamics simulations could refine binding predictions by capturing receptor flexibility and dynamic interactions.
This study underscores the critical role of structural differences between COX-1 and COX-2 in determining the selectivity and efficacy of NSAIDs. Diclofenac, a nonselective NSAID, demonstrated moderate binding affinity to COX-2 but lacked specificity, aligning with its broad-spectrum anti-inflammatory effects and higher risk of gastrointestinal and renal side effects. In contrast, etoricoxib, a selective COX-2 inhibitor, exhibited superior binding affinity and a more specific interaction profile, reducing off-target effects but raising concerns about cardiovascular safety.
The comparative analysis of docking results from AutoDock4 and AutoDock Vina highlighted the strengths and limitations of each software. While AutoDock4 provided comprehensive interaction data essential for understanding binding mechanisms, AutoDock Vina offered faster processing but limited interaction detail. This study relied on AutoDock4 for the detailed analysis of ligand-enzyme interactions, reinforcing its suitability for studies requiring precise molecular insights.
These findings provide valuable insights into the molecular basis of NSAID selectivity and efficacy, contributing to the rational design of safer and more effective anti-inflammatory agents. Future research should aim to refine the balance between effectiveness and safety in NSAID development, leveraging computational tools to optimize drug design and therapeutic strategies.
Supplementary Materials: Available online as Supplemental Material: Table S1: Values of Free Energy of Binding and Inhibition Constant for COX-2 Selective and Nonselective NSAIDs. Table S2: Average Free Binding Energy of Selective and Nonselective NSAIDs according to AutoDock4 and AutoDock Vina. Figure S1. 3D Visualization of Hydrogen Bridges between Diclofenac and COX-2 at the Active Site. Figure S2. 3D Visualization of Hydrogen Bridges between Naproxen and COX-2 at the Active Site. Figure S3. 2D Visualization of Hydrogen Bridges between Naproxen and COX-2 at the Active Site. Figure S4. 3D Visualization of Hydrogen Bridges between Ibuprofen and COX-2 at the Active Site. Figure S5. 2D Visualization of Hydrogen Bridges between Ibuprofen and COX-2 at the Active Site. Figure S6. 3D Visualization of Hydrogen Bridges between ASA and COX-2 at the Active Site. Figure S7. 2D Visualization of Hydrogen Bridges between ASA and COX-2 at the Active Site. Figure S8. 3D Visualization of Hydrogen Bridges between Etoricoxib and COX-2 at the Active Site. Figure S9. 3D Visualization of Hydrogen Bridges between Celecoxib and COX-2 at the Active Site. Figure S10. 2D Visualization of Hydrogen Bridges between Celecoxib and COX-2 at the Active Site. Figure S11. 3D Visualization of Hydrogen Bridges between Rofecoxib and COX-2 at the Active Site. Figure S12. 2D Visualization of Hydrogen Bridges between Rofecoxib and COX-2 at the Active Site.
Author Contributions: Doménica Flores: Conceptualization; methodology; software; validation; formal analysis; investigation; resources; writing—original draft preparation; writing—review and editing; visualization. Carola Jerves: supervision; methodology; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
REFERENCES
1. Martín-Vázquez E, Cobo-Vuilleumier N, López-Noriega L, Lorenzo PI, Gauthier BR. The PTGS2/COX2-PGE2 signaling cascade in inflammation: Pro or anti? A case study with type 1 diabetes mellitus. Int J Biol Sci. 2023;19(13):4157–65.
2. Salido M, Abásalo L, Bañares A. Revisión de los antiinflamatorios inhibidoresselectivos de la ciclooxigenasa-2. Inf Ter del Sist Nac Salud. 2001;25(2):46–52.
3. Rao PNP, Knaus EE. Evolution of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): Cyclooxygenase (COX) Inhibition and Beyond. J Pharm Pharm Sci [Internet]. 2008;11(2):81-110s. Available from: https://ejournals.library.ualberta.ca/index.php/JPPS/article/view/4128%0Ahttps://journals.library.ualberta.ca/jpps/index.php/JPPS/article/view/4128/3358
4. Ahmadi M, Bekeschus S, Weltmann KD, von Woedtke T, Wende K. Nonsteroidal anti-inflammatory drugs: recent advances in the use of synthetic COX-2 inhibitors. RSC Med Chem [Internet]. 2022;(February). Available from: 10.1039/d1md00280e
5. Bindu S, Mazumder S, Bandyopadhyay U. Nonsteroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem Pharmacol. 2020;180(April).
6. Peura DA, Goldkind L. Balancing the gastrointestinal benefits and risks of nonselective NSAIDs. Arthritis Res Ther [Internet]. 2005;7(SUPPL. 4):7–13. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2833976/pdf/ar1793.pdf
7. Tomić M, Micov A, Pecikoza U, Stepanović-Petrović R. Clinical Uses of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) and Potential Benefits of NSAIDs Modified-Release Preparations. Microsized Nanosized Carriers Nonsteroidal Anti-inflamm Drugs Formul Challenges Potential Benefits. 2017;1–29.
8. Muhammed MT, Aki-Yalcin E. Molecular Docking: Principles, Advances, and its Applications in Drug Discovery. Lett Drug Des Discov [Internet]. 2022;19:1–16. Available from: http://www.esisresearch.org/Uploads/Documents/esis2022muhammed(molecular-docking)lettdrugdesigndiscov.pdf
9. Novikov FN, Chilov GG. Molecular docking: theoretical background, practical applications and perspectives. Mendeleev Commun [Internet]. 2009;19(5):237–42. Available from: 10.1016/j.mencom.2009.09.001
10. Raval K, Ganatra T. Basics, types and applications of molecular docking: A review. IP Int J Compr Adv Pharmacol [Internet]. 2022;7(1):12–6. Available from: https://www.researchgate.net/profile/Keval-Raval-2/publication/359240621_Basics_types_and_applications_of_molecular_docking_A_review/links/6236ab9772d413197a33e328/Basics-types-and-applications-of-molecular-docking-A-review.pdf
11. Torrez G, Enrique R. Acomplamiento molecular: criterios prácticos para la selección de ligandos biológicamente activos e identificación de nuevos blancos terapéuticos. Rev Con-Ciencia. 2019;2:1–18.
12. Moussa N, Hassan A, Gharaghani S. Pharmacophore model, docking, QSAR, and molecular dynamics simulation studies of substituted cyclic imides and herbal medicines as COX-2 inhibitors. Heliyon [Internet]. 2021;7(4):e06605. Available from: https://doi.org/10.1016/j.heliyon.2021.e06605
13. Jara M, Jaramillo L, Matamoros JM. F recuencia de automedicación de AINES y analgésicos - antipiréticos y características que los rodean, en hogares de la parroquia San Blas de la ciudad de Cuenca en el año 2011 [Internet]. Universidad De Cuenca Facultad De Ciencias Médicas Escuela De Medicina. Universidad De Cuenc; 2011. Available from: http://dspace.ucuenca.edu.ec/bitstream/123456789/3466/1/MED95.pdf
14. Encalada C, Ortega J, Carlos Valencia. Prevalencia Y Factores Asociados a La Automedicación Con Aines En Adultos Mayores En Las Parroquias Urbanas De Cuenca [Internet]. Universidad de Cuenca. Universidad de Cuenca; 2015. Available from: http://dspace.ucuenca.edu.ec/bitstream/123456789/22494/1/tesis.pdf
15. Hanwell M, Curtis D, Lonie D, Vandermeersh T, Zurek E, Hutchison G. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;
16. Morris G, Huey R, Lindstrom W, Sanner M, Belew R, Goodsell D, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. Computational Chemistry 2009; 2009. p. 2785–91.
17. Eberhardt J, Santos-Martins D, Tillack A., Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of Chemical Information and Modeling; 2021.
18. RStudio Team. RStudio: Integrated Development for R. RStudio, PBC. 2020.
19. Schrödinger L, DeLano W. PyMOL. 2020.
20. BIOVIA DS. BIOVIA Discovery Studio [Software]. 2021.
21. Bello-Vargas E, Leyva-Peralta MA, Gómez-Sandoval Z, Ordóñez M, Razo-Hernández RS. A Computational Method for the Binding Mode Prediction of COX-1 and COX-2 Inhibitors: Analyzing the Union of Coxibs, Oxicams, Propionic and Acetic Acids. Pharmaceuticals. 2023;16(12).
22. Chaudhary N, Aparoy P. Deciphering the mechanism behind the varied binding activities of COXIBs through Molecular Dynamic Simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies. J Biomol Struct Dyn. 2017;35(4):868–82.
23. Ibrahim MM, Elsaman T, Al-Nour MY. Synthesis, Anti-Inflammatory Activity, and In Silico Study of Novel Diclofenac and Isatin Conjugates. Int J Med Chem. 2018;2018:1–11.
24. Meneses L, Cuesta S. Determinación Computacional de la Afinidad y Eficiencia de Enlace de Antinflamatorios No Esteroideos Inhibidores de la Ciclooxigenasa-2. Rev Ecuat Med Cienc Biol. 2015;36(1–2):17–25.
25. Sahu MK, Nayak AK, Hailemeskel B, Eyupoglu OE. Exploring Recent Updates on Molecular Docking: Types, Method, Application, Limitation & Future Prospects. Int J Pharm Res Allied Sci. 2024;13(2):24–40.
26. Tobi D, Bahar I. Optimal design of protein docking potentials: Efficiency and limitations. Proteins Struct Funct Genet. 2006;62(4):970–81.
Received: February 7, 2025 / Accepted: March 28.2025 / Published: June 15, 2025
Citation: Flores D, Jerves C. Computational Comparison of the Binding Affinity of Selective and Nonselective NSAIDs to COX-2 Using Molecular Docking. Bionatura journal. 2025;2 (2):3. doi: 10.70099/BJ/2025.02.02.3
Additional information Correspondence should be addressed: carola.jerves@ucuenca.edu.ec
Peer review information. Bionatura thanks anonymous reviewer(s) for their contribution to the peer review of this work using https://reviewerlocator.webofscience.com/
ISSN. 3020-7886
All articles published by Bionatura Journal are made freely and permanently accessible online immediately upon publication, without subscription charges or registration barriers.
Publisher's Note: Bionatura Journal stays neutral concerning jurisdictional claims in published maps and institutional affiliations.
Copyright: © 2025 by the authors. They were submitted for possible open-access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).