Critical Mini-Review
Beyond the ATP Pocket: Critical Analysis of Substrate-Directed CK2 Inhibition in Oncology, Virology, and Neurodegeneration

Nelson Santiago Vispo  1*
1*
1 Clinical Biotec SL. and Bionatura Journal. Madrid. 28029. Spain. Member of the Scientific Board, Bionatura Journal (role declared for transparency only).
*Corresponding author. santiago@clinicalbiotec.com
ABSTRACT
Protein kinase CK2 is a constitutively active serine/threonine kinase that regulates cell proliferation, survival, and stress responses. Dysregulation of CK2 is implicated in oncology, virology, and neurodegeneration, establishing it as a significant therapeutic target. While initial efforts focused on ATP-competitive inhibitors such as silmitasertib, recent strategies have investigated modulation outside the catalytic pocket, including substrate-directed inhibition. The clinical peptide CIGB-300 exemplifies this approach. Although originally designed to block phosphoacceptor motifs on CK2 substrates, subsequent evidence indicates a multimodal mechanism involving direct interaction with the catalytic subunit. This mini-review critically evaluates the transition toward non-ATP-competitive strategies and examines translational evidence for CK2 modulation in oncology, virology, and neurodegeneration. The analysis demonstrates that oncologic applications possess the most robust preclinical and clinical validation, whereas evidence in virology and neurodegeneration remains primarily preliminary or mechanistic. The review distinguishes between the maturity of evidence in these fields and its implications for guiding future research.
Keywords: CK2 inhibition; Substrate-directed therapy; CIGB-300; Oncology; Virology; Neurodegeneration.
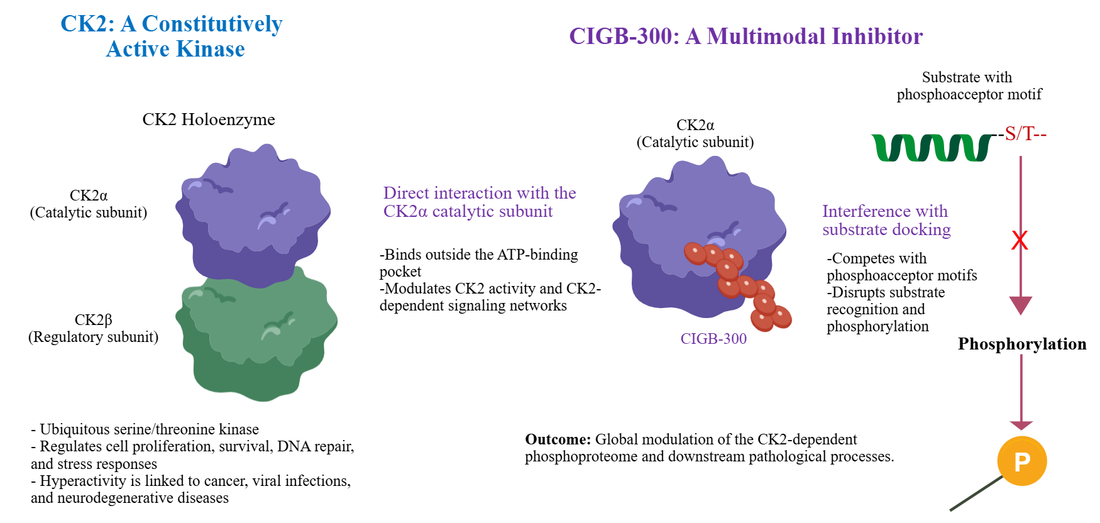
Graphical Abstract. Schematic representation of the transition from constitutive CK2 activity to multimodal inhibition by CIGB-300. (Left) The CK2 holoenzyme, comprising the catalytic (CK2α) and regulatory (CK2β) subunits, functions as a constitutively active serine/threonine kinase driving pathological processes. (Center) CIGB-300 acts as a multimodal inhibitor, directly interacting with the CK2α catalytic subunit. (Right) This interaction competitively blocks substrate docking and prevents phosphorylation at phosphoacceptor motifs (S/T), ultimately modulating the global CK2-dependent phosphoproteome and downstream disease mechanisms.
INTRODUCTION
Protein kinase CK2 is a ubiquitous, constitutively active enzyme involved in cell cycle control, inhibition of apoptosis, DNA repair, and transcriptional regulation. Due to its pleiotropic roles, CK2 hyperactivity is a common feature in diverse pathologies, particularly solid and hematologic malignancies, viral infections, and neurodegenerative disorders. However, the ubiquity of CK2 poses a therapeutic challenge: systemic inhibition risks disrupting essential physiological processes and triggering compensatory mechanisms 1.
For decades, pharmacological inhibition primarily relied on ATP-competitive compounds targeting the highly conserved catalytic pocket. Molecules such as silmitasertib (CX-4945) validated CK2 as a druggable target but also highlighted limitations in selectivity and the risk of on-target toxicity due to the kinase's broad physiological functions. As a result, alternative strategies designed to modulate specific CK2 interactions, including allosteric modulators, holoenzyme disruptors, and substrate-directed inhibitors, have gained increasing attention 2.
Among these approaches, the peptide CIGB-300 represents one of the most clinically advanced examples of non-ATP-competitive inhibition. Initially designed to bind the phosphoacceptor domain of CK2 substrates, its mechanism has been re-evaluated to include direct interactions with the CK2α catalytic subunit 3-7. This mini-review analyzes the evolution of CK2 targeting strategies, using CIGB-300 as a case study to assess the translational maturity of CK2 inhibition in oncology, virology, and neurodegeneration.
From ATP-Competitive To Substrate-Directed Inhibition
Classical CK2 inhibition targets the ATP-binding site through orthosteric inhibition. Although effective in preclinical models, the structural conservation of this site across the kinome raises concerns regarding specificity and functional redundancy within cellular signaling networks 2. In response, substrate-directed strategies have emerged to block the interaction between CK2 and specific pathological substrates without globally suppressing kinase activity. In response to these limitations, the pharmacological targeting of CK2 has expanded beyond the orthosteric site to explore allosteric modulation and holoenzyme disruption 1,2. Because CK2 typically functions as a tetrameric complex (CK2α₂β₂), targeting the protein–protein interface between the catalytic (CK2α/α′) and regulatory (CK2β) subunits offers a theoretical mechanism to alter substrate recognition and subcellular localization without competing with high intracellular ATP concentrations 2. Similarly, the identification of cryptic allosteric pockets distinct from the active site has opened avenues for developing modulators that could provide enhanced selectivity for specific CK2 isoforms (e.g., CK2α versus CK2α′), a feature highly desirable for mitigating on-target toxicity in non-malignant tissues 1,2. While these structural approaches are mechanistically elegant, translating them into clinically viable drugs remains challenging due to issues with cell permeability and target engagement. It is precisely within this broader landscape of 'outside the catalytic box' strategies that substrate-competitive approaches must be contextualized.
CIGB-300 was rationally designed to target the CK2 phosphorylation site on the HPV-16 E7 oncoprotein, based on the identification of cyclic peptides that bind this specific motif 3,4. This substrate-competitive mechanism theoretically offered improved selectivity. However, proteomic and phosphoproteomic studies have challenged this binary perspective. Current evidence demonstrates that CIGB-300 interacts with both substrates and the CK2α catalytic subunit, modulating the broader CK2-dependent phosphoproteome 5-7. Therefore, CIGB-300 is more accurately classified as a multimodal inhibitor rather than a purely substrate-directed agent. Recognizing this distinction is essential for understanding its pharmacological properties and potential toxicities, rather than relying on the simplified model of ATP-site competition.
Oncology
The association between CK2 and tumorigenesis is well established, involving the promotion of proliferation, the inhibition of apoptosis, and the facilitation of metastasis 1. Consequently, oncology remains the field with the most substantial evidence supporting CK2 as a therapeutic target.
For CIGB-300, the data are most robust in this domain. Initial studies demonstrated proapoptotic effects in cervical cancer models linked to the disruption of the HPV E7 oncoprotein interaction with the retinoblastoma (RB) complex 3,6. The rationale was strongly supported by the first patent, which identified peptides capable of sensitizing tumors to interferon and blocking E7 phosphorylation 3. Subsequent clinical trials and proteomic analyses demonstrated that CIGB-300 impacts CK2-dependent signaling networks, reducing tumor viability in vivo 4,5,7.
Oncology thus serves as the primary context for the development of CK2 inhibitors. The transition from the mechanistic hypothesis of substrate blocking to clinical observations of broad modulation of the phosphoproteome is most thoroughly documented in this field, providing a strong foundation for therapeutic application.
Virology
The role of CK2 in viral infections, particularly SARS-CoV-2, has attracted significant interest. Phosphoproteomic analyses have shown that SARS-CoV-2 infection hijacks CK2 to facilitate cytoskeletal remodeling and filopodia formation, processes necessary for viral egress 8. These findings position CK2 as a promising host-directed target, potentially offering a higher barrier to viral resistance than direct-acting antivirals.
In this context, CIGB-300 was repositioned for the treatment of COVID-19. Early-phase clinical trials suggested improvements in radiological parameters and inflammatory biomarkers in hospitalized patients 9. Furthermore, preclinical studies in bovine coronavirus models showed antiviral efficacy 10.
However, a critical appraisal is necessary. While the mechanistic rationale for targeting CK2 in coronaviruses is strong, the clinical data for CIGB-300 in this indication remain preliminary compared with those of established antivirals targeting viral proteins (e.g., Mpro). Current evidence supports its capacity as a complementary host-directed therapy rather than a standalone standard of care at this stage 8-11.
Neurodegeneration
Mechanistic Convergence With Limited Translational Data
In neurodegenerative diseases, CK2 activity has been linked to tau pathology and synaptic dysfunction. In Alzheimer’s disease models, CK2 phosphorylates Tau and regulates NMDA receptor localization, and inhibition attenuates pathological markers 12. Similarly, in Huntington’s disease, the CK2α subunit influences neuroinflammation and synaptic integrity 13.
Despite this biological plausibility, the application of CIGB-300 or similar CK2 modulators in neurodegenerative disease remains speculative. The current literature provides a mechanistic rationale for CK2's involvement but lacks direct translational evidence linking CIGB-300 to neurological outcomes. Unlike oncology, where the translational pipeline is active, neurodegeneration represents a future opportunity for CK2-targeted therapies, contingent on the development of molecules that can cross the blood-brain barrier and demonstrate chronic safety 12,13.
Limitations Of The Current Evidence
While the therapeutic potential of CK2 modulation is evident, several limitations have to be acknowledged. First, the evidence base is heterogeneous: it is robust in oncology but remains largely preclinical and preliminary in virology and neurodegeneration, limiting direct extrapolation across indications. Second, pharmacokinetic challenges associated with peptide-based inhibitors like CIGB-300 (e.g., metabolic stability, tissue penetration, and delivery) may limit their broad clinical application compared with small-molecule ATP-competitive inhibitors. Finally, although multimodal inhibition may offer therapeutic advantages, the precise in vivo contributions of substrate binding versus catalytic subunit interactions remain incompletely understood, and a comprehensive characterization of potential off-target effects, particularly under chronic dosing, is lacking.
Second, the inherent pharmacokinetic challenges associated with peptide-based inhibitors—such as rapid metabolic degradation, limited systemic tissue penetration, and the initial reliance on intratumoral injection routes—pose significant hurdles to broad clinical application compared with small-molecule ATP-competitive inhibitors. This pharmacological complexity partly explains why CIGB-300, despite demonstrating consistent biological activity and safety over nearly two decades, has not yet advanced to pivotal Phase III oncology trials. However, rather than indicating a stalled pipeline, current evidence suggests a strategic shift toward resolving these delivery barriers. Recent research is actively investigating CIGB-300’s efficacy using advanced 3D tumoroid models that better mimic in vivo complexity, as well as exploring its interplay with tumor metabolism (e.g., FASN) and immune alarmins to design rational combination therapies 14-16.
Second, the inherent pharmacokinetic challenges associated with peptide-based inhibitors—such as rapid metabolic degradation, limited systemic tissue penetration, and the initial reliance on intratumoral injection routes—pose significant hurdles to broad clinical application compared with small-molecule ATP-competitive inhibitors. Nonetheless, developing appropriate nanoformulations of peptide-based drugs represents a promising strategy to overcome the vulnerability of plain peptides in blood. However, rather than indicating a stalled pipeline, current evidence suggests a strategic shift toward resolving these delivery barriers. Recent research is actively investigating CIGB-300’s efficacy using advanced 3D tumoroid models that better mimic in vivo complexity, as well as exploring its interplay with tumor metabolism (e.g., FASN) and immune alarmins to design rational combination therapies 14-16.
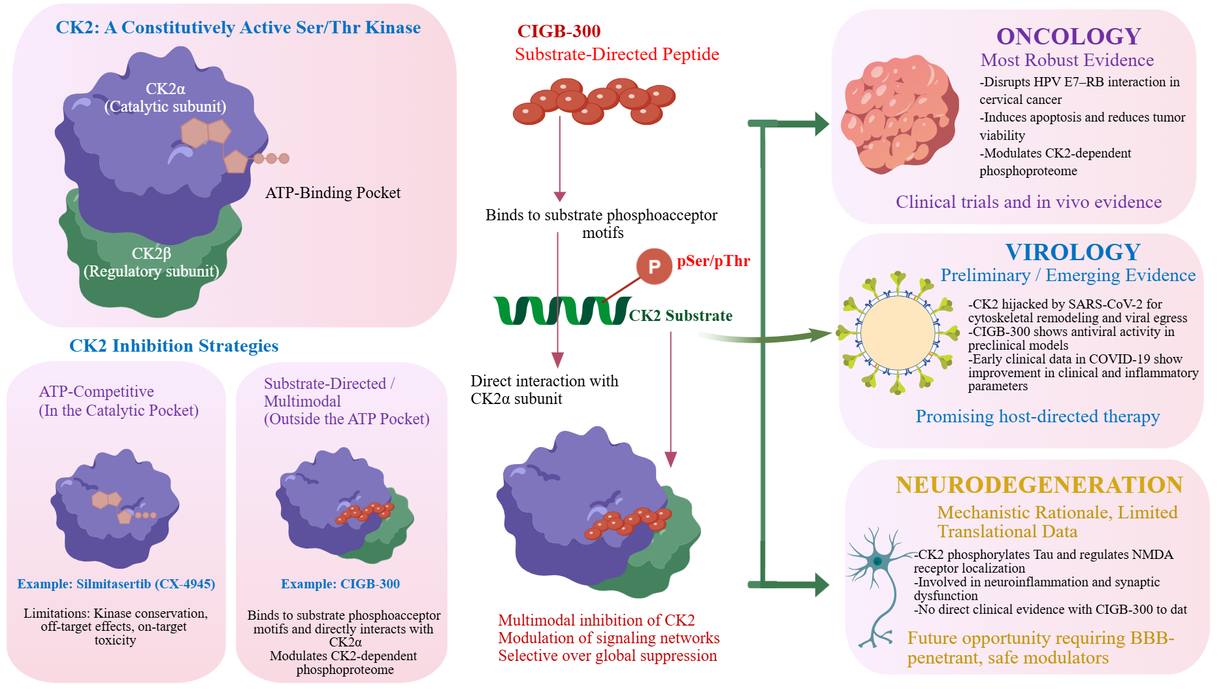
Figure 1. Mechanistic evolution and translational maturity of CK2 inhibition strategies. (Top panel) Contrasting mechanisms of action: Left, orthosteric ATP-competitive inhibition by small molecules (e.g., silmitasertib/CX-4945) blocking the catalytic pocket (PDB: 7L1X); Right, multimodal inhibition by the CIGB-300 peptide, which concurrently binds to the CK2α catalytic subunit and the phosphoacceptor domains of specific substrates (e.g., HPV-16 E7), disrupting broader CK2-dependent protein-protein interaction networks. (Bottom panel) Hierarchical map of current translational evidence across pathologies. Oncology exhibits the highest maturity, supported by clinical trials and phosphoproteomic mapping of the E7-RB axis disruption 5,7. Virology shows preliminary clinical potential as a host-directed therapy targeting CK2-mediated cytoskeletal remodeling during SARS-CoV-2 egress 8,9. Neurodegeneration remains at a mechanistic stage, with a primary focus on linking CK2 activity to Tau pathology and synaptic NR2B mislocalization 12.
Future Directions
Future research should prioritize three key areas. First, the development of non‑peptidic mimetics that preserve the selectivity of substrate‑directed CK2 inhibition while improving pharmacokinetic properties and overall bioavailability. Second, the design and characterization of subunit‑selective CK2 modulators (e.g., preferentially targeting CK2α′ in neurodegenerative settings) to more precisely fine‑tune therapeutic effects and safety profiles. Finally, well‑powered clinical trials in virology, together with rigorous preclinical studies in neurodegeneration, are needed to validate the cross‑disease utility of CK2 modulation beyond oncology.
CONCLUSIONS
CK2 represents a transversal therapeutic target with validated importance in oncology and emerging roles in virology and neurodegeneration. The development of CIGB-300 illustrates the evolution from ATP-competitive inhibition to complex, multimodal strategies that target kinase-substrate interactions. However, the translational maturity of this approach varies considerably across disease areas. Oncology currently provides the strongest support for clinical application, while applications in virology and neurodegeneration, though mechanistically promising, require more rigorous validation. Distinguishing between these levels of evidence is critical for the rational advancement of CK2-targeted therapies.
Author Contributions: N.S.V. is the sole author of this review, responsible for the conceptualization, literature analysis, drafting, and final revision of the manuscript.
Ethical Statement and Editorial Independence: The author of this manuscript serves as the Editor-in-Chief of Bionatura Journal. To ensure the integrity of the publication process and prevent editorial bias, this article was handled independently by an Associate/Guest Editor. The peer-review process was conducted in a double-blind manner by external experts, and the author had no involvement in reviewer selection or final decision-making.
Conflicts of Interest: The author has completed the ICMJE uniform disclosure form. The author declares a historical professional involvement in the early development and characterization of the CIGB-300 peptide (formerly known as P15-Tat) during the early 2000s, as documented in the cited literature (References 3 and 4). The author is currently affiliated with Clinical Biotec SL. This review was conducted with academic independence, and no direct or indirect financial support was received from the developers of the molecules discussed in this article.
Funding: This research received no external funding.
Institutional Review Board Statement: Not applicable. This article is a critical mini-review based on publicly available literature and does not involve any new studies with human participants or animals performed by the author.
Informed Consent Statement: Not applicable.
Data Availability Statement: All data generated or analyzed during this review are publicly available in the cited references and their corresponding databases.
Acknowledgments: Not applicable.
AI-Assisted Tools Disclosure: During the initial conceptualization phase, the AI tool GPAI (https://gpai.app/) was used solely to explore preliminary visual layouts and compositional ideas. No artificial intelligence system was used to generate, manipulate, or analyze experimental data, clinical data, statistical results, or scientific conclusions. To ensure scientific accuracy and comply with BioNatura Journal policies, the final figures were manually reconstructed, scientifically verified, and rendered by the author using BIOGDP. The author independently reviewed and verified the final content, interpretations, and conclusions, in compliance with the BioNatura Journal policy: https://bionaturajournal.com/artificial-intelligence--ai-.html
REFERENCES
1. Borgo Christian, D'Amore Claudio, Sarno Silvia, Salvi Mauro, Ruzzene Maria. Protein kinase CK2: a potential therapeutic target for diverse human diseases. Signal Transduction and Targeted Therapy. 2021;6(1):183. Disponible en: https://doi.org/10.1038/s41392-021-00567-7
2. Day-Riley Sarah, Gray Nathanael, Bullock Alex N. CK2 Inhibitors Targeting Inside and Outside the Catalytic Box. Kinases and Phosphatases. 2024;2(2):101-121. Disponible en: https://doi.org/10.3390/kinasesphosphatases2020007
3. Perea Rodríguez Silvio Ernesto, Reyes Acosta Osvaldo, Santiago Vispo Nelson Francisco, Puchades Izaguirre Yanier, Silva Rodríguez Ricardo, Moro Soria Alejandro, Santos Savio Alicia, González López Luis Javier, Castellanos Serra Lila Rosa, Musacchio Lasa Alexis, Gil Valdés Jeovanis, Rodríguez Rodríguez Leticia, Gerónimo Pérez Hermis, Besada Pérez Vladimir Armín, González Basterrechea Leonor, Padrón Palomares Gabriel Ramón, inventores; Centro de Ingeniería Genética y Biotecnología, solicitante. Peptides for the treatment of cancer associated with the human papilloma virus (HPV) and other epithelial tumours. Patente Internacional WO 03/054002 A1. 2003 Jul 3.
4. Perea Silvio E, Reyes Osvaldo, Puchades Yanier, Mendoza Orlando, Vispo Nelson S, Torrens Idania, Santos Alicia, Silva Ricardo, Lukaszewicz Beata, Szatkowski Krzysztof, Troszczynska Maria, Castellanos Lila, Musacchio Alexis, Gil Jeovanis, Rodríguez Leticia, Gerónimo Hermis, Besada Vladimir, González Leonor, Padrón Gabriel R. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Research. 2004;64(19):7127-7129. Disponible en: https://doi.org/10.1158/0008-5472.CAN-04-1210
5. Perera Yasser, Costales Adriana, Díaz-Rodríguez Yanisley, García-Castillo Idania, Reyes Osvaldo, Perea Silvio E. CIGB-300 Anticancer Peptide Regulates the Protein Kinase CK2-Dependent Phosphoproteome. Pharmaceutics. 2020;12(6):525. Disponible en: https://doi.org/10.3390/pharmaceutics12060525
6. Ramón Ana C, Bringas Ricardo, Perera Yasser, Perea Silvio E. CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma Complex in Cervical Cancer Cells. Viruses. 2022;14(8):1681. Disponible en: https://doi.org/10.3390/v14081681
7. Pérez Gretel V, Perera Yasser, Bringas Ricardo, Ramón Ana C, Perea Silvio E. CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model. Biomedicines. 2023;11(1):43. Disponible en: https://doi.org/10.3390/biomedicines11010043
8. Bouhaddou Mehdi, Memon Danish, Meyer Benjamin, White Kris M, Rezelj Veronica V, Correa Marrero Marisa, Polacco Benjamin J, Melnyk Jennifer E, Ulferts Sascha, Kaake Robyn M, Batra Jyoti, Zheng Alan L, Xu Ke, Bouhaddou Mohamed, Obernier Kirsten, Fabius Monique, Guillén Jeanette, Swaney Danielle L, Rosales David L, Tuttle Thomas G, Richardson Elizabeth S, Aneja Jneesh, Shah Arpit S, Gopal Sreejith, Choudhary Arpit, Shokat Kevan M, García-Sastre Adolfo, Vignuzzi Marco, Johnson Jeffrey R, Krogan Nevan J. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182(3):685-712.e19. Disponible en: https://doi.org/10.1016/j.cell.2020.06.034
9. Cruz Luis R, Baladrón Idania, Rittoles Alis, Díaz Yanisley, Perera Yasser, Resik Sonia, Castellanos María J, González Dania, Fernández Mayra, García Idania, Pérez Gretel V, Ramón Ana C, Valenzuela Carmen, Hernández-Bernal Francisco, Abdo-Cuza Alejandro, Brown-Govea Alberto, González-García José L, Morera-Valles Manuel, López-Sánchez Idelsy, Perea Silvio E. Treatment with an Anti-CK2 Synthetic Peptide Improves Clinical Response in COVID-19 Patients with Pneumonia. A Randomized and Controlled Clinical Trial. ACS Pharmacology & Translational Science. 2021;4(1):206-212. Disponible en: https://doi.org/10.1021/acsptsci.0c00174
10. Ramón Ana C, Bringas Ricardo, Perera Yasser, Perea Silvio E. Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection. Viruses. 2022;14(3):552. Disponible en: https://doi.org/10.3390/v14030552
11. Quezada Meza Claudia P, Ruzzene Maria. Protein Kinase CK2 and SARS-CoV-2: An Expected Interplay Story. Kinases and Phosphatases. 2023;1(2):141-150. Disponible en: https://doi.org/10.3390/kinasesphosphatases1020010
12. Marshall Courtney A, McBride J David, Changolkar Laxmi, Riddle Adam, Trojanowski John Q, Lee Virginia M-Y. Inhibition of CK2 mitigates Alzheimer's tau pathology by preventing NR2B synaptic mislocalization. Acta Neuropathologica Communications. 2022;10(1):30. Disponible en: https://doi.org/10.1186/s40478-022-01333-z
13. Yu Di, Zhang Yang, Wang Hong, Zhang Zhentao, Liu Jun, Wang Jialing, Wang Xiaofeng, Chen Jianfeng, Gu Xiaosong, Wang Xiaochun. CK2 alpha prime and alpha-synuclein pathogenic functional interaction mediates synaptic dysregulation in Huntington's disease. Acta Neuropathologica Communications. 2022;10(1):83. Disponible en: https://doi.org/10.1186/s40478-022-01385-1
14. Paduano G, Stefanizzi V, Garay H, Perea SE, et al. Microenvironment Rheology Modulates the Effect of the Anticancer Peptide CIGB-300 on 3D Head and Neck Tumoroids. Int J Mol Sci. 2026;27(4):1973. doi: 10.3390/ijms27041973
15. Jiang X, Fang G, Li W, Liu Y, Chen G, Perea SE, et al. FASN at the Crossroads of Tumor Metabolism, Immune Evasion, and Therapy Resistance: Mechanistic Insights and Therapeutic Opportunities. Crit Rev Oncol Hematol. 2026;194:104231. doi: 10.1016/j.critrevonc.2025.104231
16. Valenzuela C, Zhao Q, Zhang Z, Li W, Perea SE, et al. Circulating levels of high mobility group box-1 and nucleophosmin/B23 proteins and clinical significance in debut non-small cell lung cancer patients. Sci Rep. 2026;16:4512. doi: 10.1038/s41598-025-58214-x
Received: March 02, 2026 / Accepted: May 10, 2026 / Published (Online First): May 12, 2026 / Issue Date: June 15, 2026 (Europe/Madrid)
Citation: Vispo NS. Beyond the ATP Pocket: Critical Analysis of Substrate-Directed CK2 Inhibition in Oncology, Virology, and Neurodegeneration – A Critical Mini-Review. BioNatura Journal: Ibero-American Journal of Biotechnology and Life Sciences. 2026;3(2):5. https://doi.org/10.70099/BJ/2026.03.02.5
Correspondence should be addressed to: santiago@clinicalbiotec.com
Peer Review Information: BioNatura Journal thanks the anonymous reviewers for their valuable contribution to the peer-review process. Regional peer-review coordination was conducted under the BioNatura Institutional Publishing Consortium (BIPC). Reviewer selection and assignment were supported via: https://www.reviewercredits.com/
Digital Preservation and Repository: This journal is managed through the Open Journal Systems (OJS) platform. To ensure long-term access, we use the PKP Preservation Network (PKP PN) to digitally preserve all published volumes in a decentralized, secure archive. Furthermore, our repository is integrated with LOCKSS and CLOCKSS, allowing international library networks to create permanent archives for long-term survival.
Publisher Information: Published by Clinical Biotec S.L. (Madrid, Spain) as the publisher of record under the BioNatura Institutional Publishing Consortium (BIPC). Places of publication: Madrid (Spain); Tegucigalpa (Honduras); Panama City (Panama). Online ISSN: 3020-7886.
BioNatura Publishing Consortium (BIPC) © Clinical Biotec S.L. – Madrid, Spain