ANÁLISIS DEL COMPLEJO TIROSINASA-ÁCIDO PROTOCATECUICO MEDIANTE DOCKING Y DINÁMICA MOLECULAR
Oscar A. Pineda-Maldonado1, Alejandro J. Cuevas-Martínez1, Oscar V. Ortiz-Hernández1, Jorge M. Vargas-Zuniga1, Evelina D. Estrada-López1*
1Grupo de Investigación CyTMAE, Departamento de Ingeniería Química, UNAH, Tegucigalpa, Honduras.
Autor correspondiente: evelina.estrada@unah.edu.hn
INTRODUCCIÓN
El oscurecimiento de la mayoría de los vegetales es un proceso natural que afecta negativamente su valor comercial y la aceptación por parte de los consumidores. Este fenómeno, conocido como pardeamiento enzimático, es causado por la acción de la enzima tirosinasa (1). La tirosinasa es responsable de catalizar la oxidación de los fenoles presentes en estos vegetales, convirtiéndolos en quinonas, las cuales se visualizan como pigmentos oscuros, deteriorando la apariencia de los productos vegetales (2). Alimentos como los champiñones, papas, aguacates, coliflor, berenjenas y manzanas son especialmente vulnerables a este fenómeno de pardeamiento enzimático. La comprensión de este proceso y la búsqueda de soluciones efectivas son esenciales para mantener la calidad y la apariencia de los productos vegetales frescos (3).
Los compuestos fenólicos muestran potencial como inhibidores naturales de la tirosinasa; muchos de estos se encuentran en subproductos del sector agroindustrial, como la pulpa del café (4). El ácido protocatecuico, presente en la pulpa de café hondureño, podría ser un potencial inhibidor de la tirosinasa (PCA) (5,6). La inhibición de la tirosinasa por el PCA puede estudiarse mediante diversas metodologías experimentales; sin embargo, las técnicas computacionales han demostrado ser una herramienta poderosa para comprender estos procesos a nivel molecular.
El docking molecular es un método computacional cuyo objetivo es predecir las conformaciones predominantes entre una proteína de estructura conocida y otra estructura química llamada ligando. Este método permite estimar la energía de enlace, lo que resulta útil para determinar el complejo proteína-ligando más estable y predecir el sitio activo de una enzima (7). Por otro lado, la dinámica molecular (MD) es otro método computacional que busca predecir cómo evoluciona la estructura de una macromolécula en el tiempo. Asimismo, es una herramienta que permite analizar los cambios estructurales que sufre una macromolécula cuando interactúa con otras moléculas (8).
Con esta investigación se pretende combinar estas dos técnicas computacionales para conocer el mejor complejo y su sitio activo, así como el mecanismo de interacción a nivel molecular de la enzima-ligando a lo largo del tiempo, con ayuda de los análisis de desviación media cuadrática (RMSD) y radio de giro como medidas de estabilidad y plegamiento.
METODOLOGÍA
La estructura de la proteína tirosinasa del hongo Agaricus bisporus (PDB ID: 2y9x) fue recuperada de Protein Data Bank y la geometría del ácido protocatecuico fue obtenida de PubChem. Utilizando AutoDock v4.2.6 y AutoDock Tools v1.5.7(9) como interfaz gráfica, se realizó un análisis de las interacciones entre la tirosinasa y el PCA con residuos rígidos, permitiendo la torsión de los átomos rotables del ligando. Se definieron las coordenadas de búsqueda en X = -4.773, Y = -8.683 y Z = -68.38, con un tamaño de 126 × 110 × 126 puntos y un espaciado de 1.2 Å. Utilizando un algoritmo lamarckiano, se determinó la conformación con la menor energía de enlace.
Las dinámicas moleculares clásicas, tanto de la tirosinasa sola como del complejo, fueron realizadas con el paquete computacional GROMACS 2023(10) y el campo de fuerza OPLS-AA (11). Las estructuras fueron colocadas en una caja con agua y neutralizadas. El campo de fuerza de la proteína fue generado con el programa gmx pdb2gmx de GROMACS, mientras que el campo de fuerza para el ácido se generó con LigParGen(12). Los sistemas fueron minimizados en sus energías usando el algoritmo de steepest descent(13). Se realizó un equilibrio por separado, tanto de temperatura como de presión, con ensembles NVT/NPT a 300 K y 1 bar durante 200 ps. Posteriormente, se realizó una dinámica molecular de 5 ns utilizando el algoritmo leap frog(14).
RESULTADOS
En la tabla 1 se muestran las energías de enlace del complejo, siendo las conformaciones 9 y 10 las que presentan una menor energía de enlace de -2.05 kcal mol⁻¹ y mayor estabilidad. Esta estructura del complejo fue utilizada para realizar la dinámica molecular.
En la ilustración 1 se pueden observar las interacciones del ligando con los residuos de la tirosinasa en el sitio activo. El grupo amino de la lisina (LYS1190) forma un enlace con el grupo hidroxilo del ligando. Además, se generan tres interacciones hidrofóbicas con los siguientes residuos: glicina (GLY1187), lisina (LYS1301) y ácido aspártico (ASP1302).
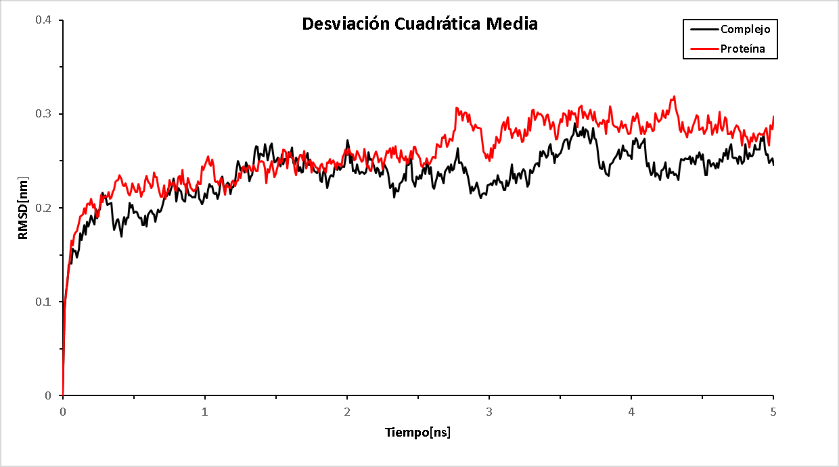
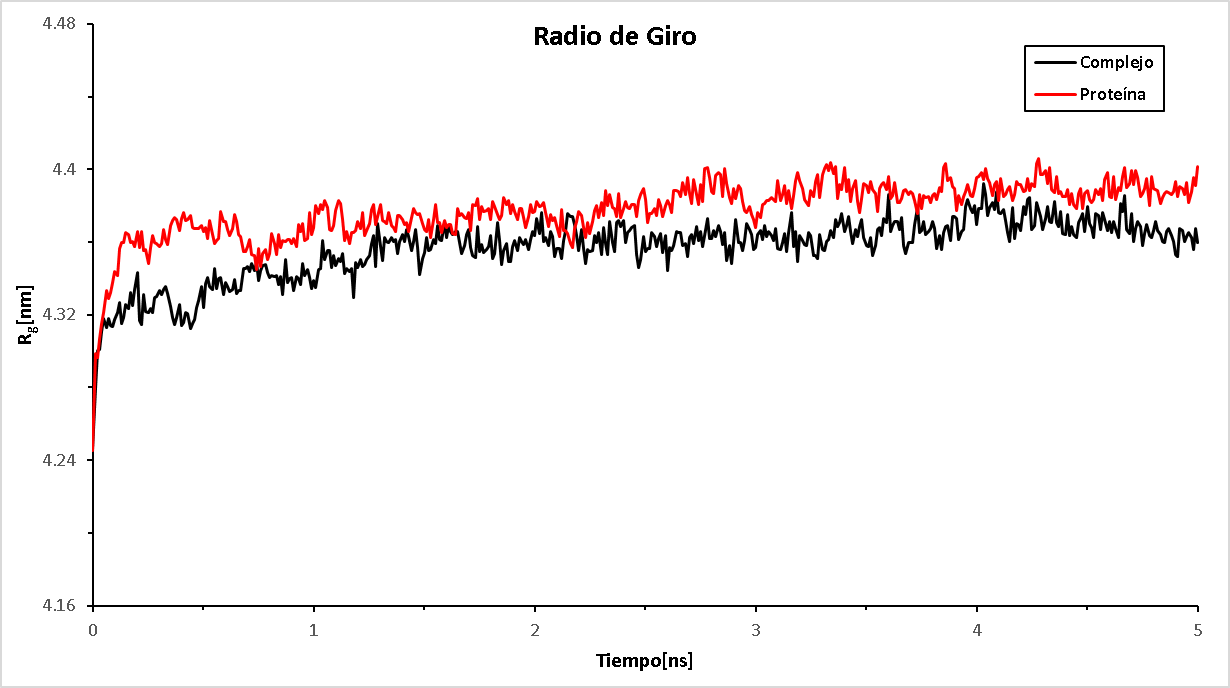
En la ilustración 3 se muestra que tanto la enzima como el complejo presentan una RMSD estable a partir de los 3 ns de simulación, lo que indica que los análisis realizados después de ese tiempo serán estables. En la ilustración 4 se presentan los resultados del radio de giro de la enzima y del complejo durante todo el tiempo de simulación. El complejo obtuvo menores valores de radio de giro, lo que indica que el ligando produce un efecto de mayor plegamiento en la enzima.
Conformación | Energía de Enlace [kcal mol-1] |
9 y 10 | -2.05 |
1 | -1.65 |
2 | -1.48 |
5 | -1.40 |
7 | -1.39 |
4 | -1.21 |
8 | -1.19 |
6 | -1.18 |
3 | -0.93 |
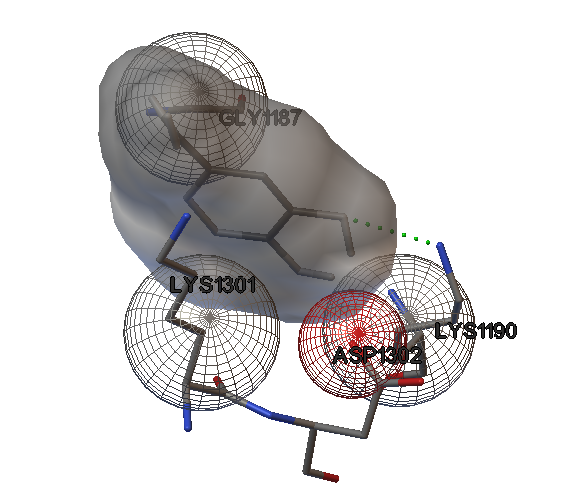
Tabla 1. Energías de enlace en kcal mol-1 para el complejo. Ilustración 1. Sitio activo del complejo.
Ilustración 2. Desviación cuadrática media de la enzima y el complejo en 5 ns. |
Ilustración 3. Radio de giro de la enzima y el complejo en un tiempo de 5 ns. |
CONCLUSIÓN
Se logró realizar el estudio de docking y dinámica computacional durante 5 ns. La enzima mostró un enlace con el ligando, y el complejo presentó estabilidad y mayor plegamiento durante el tiempo de simulación, lo que indica que el PCA podría ser utilizado como inhibidor del proceso de oxidación de vegetales. Sin embargo, para reafirmar estos resultados, es necesario realizar estudios de docking con átomos flexibles, incrementar el tiempo de simulación computacional y llevar a cabo estudios experimentales.
REFERENCIAS
1. Dergal SB. Química de los Alimentos. Sexta. México: Pearson; 2020.
2. YORUK R, MARSHALL MR. PHYSICOCHEMICAL PROPERTIES AND FUNCTION OF PLANT POLYPHENOL OXIDASE: A REVIEW. J Food Biochem [Internet]. 2003 Nov;27(5):361–422. Available from: https://onlinelibrary.wiley.com/doi/10.1111/j.1745-4514.2003.tb00289.x
3. Tilley A, McHenry MP, McHenry JA, Solah V, Bayliss K. Enzymatic browning: The role of substrates in polyphenol oxidase mediated browning. Curr Res Food Sci [Internet]. 2023;7:100623. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2665927123001910
4. Shojazadeh T, Zolghadr L, Gharaghani S, JafarKhani S, Molaabasi F, Piri H, et al. New insights into the inhibitory effect of phenol carboxylic acid antioxidants on mushroom tyrosinase by molecular dynamic studies and experimental assessment. J Biomol Struct Dyn [Internet]. 2023 Dec 29;41(22):13404–14. Available from: https://www.tandfonline.com/doi/full/10.1080/07391102.2023.2175038
5. Bulnes D, Melgar S, Vega E, Rubio A, Espinal A, Velásquez-Tinoco DG, et al. A look into Honduran biomass: facts, uses and potential applications. Bionatura [Internet]. 2023 Sep 15;8(3):1–18. Available from: https://www.revistabionatura.com/2023.08.03.37.html
6. Chen C-Y, Shih C-H, Lin T-C, Zheng J-H, Hsu C-C, Chen K-M, et al. Antioxidation and Tyrosinase Inhibitory Ability of Coffee Pulp Extract by Ethanol. Cilindre C, editor. J Chem [Internet]. 2021 Dec 17;2021:1–8. Available from: https://www.hindawi.com/journals/jchem/2021/8649618/
7. Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc [Internet]. 2016 May 14;11(5):905–19. Available from: https://www.nature.com/articles/nprot.2016.051
8. Hollingsworth SA, Dror RO. Molecular Dynamics Simulation for All. Vol. 99, Neuron. Cell Press; 2018. p. 1129–43.
9. Fu Y, Zhao J, Chen Z. Insights into the Molecular Mechanisms of Protein-Ligand Interactions by Molecular Docking and Molecular Dynamics Simulation: A Case of Oligopeptide Binding Protein. Comput Math Methods Med. 2018;2018.
10. Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25.
11. Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B [Internet]. 2001 Jul 1;105(28):6474–87. Available from: https://doi.org/10.1021/jp003919d
12. Dodda LS, Cabeza de Vaca I, Tirado-Rives J, Jorgensen WL. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res [Internet]. 2017 Jul 3;45(W1):W331–6. Available from: https://doi.org/10.1093/nar/gkx312
13. Alexander ST. The Method of Steepest Descent. Adapt Signal Process. 1986;46–67.
14. Taylor P, Gunsteren WF Van, Berendsen HJC. A Leap-frog Algorithm for Stochastic Dynamics A LEAP-FROG ALGORITHM FOR STOCHASTIC DYNAMICS. 2007;(May 2013):37–41.
Cómo citar este trabajo (Vancouver):
Pineda-Maldonado OA, Cuevas-Martínez AJ, Ortiz-Hernández OV, Vargas-Zuniga JM, Estrada-López ED. ANÁLISIS DEL COMPLEJO TIROSINASA-ÁCIDO PROTOCATECUICO MEDIANTE DOCKING Y DINÁMICA MOLECULAR [resumen]. En: Vispo NS, editor. Memorias del Congreso de Investigación y Posgrado UNAH 2024: Libro de resúmenes. Madrid/Tegucigalpa: Clinical Biotec S.L.; Universidad Nacional Autónoma de Honduras; 2024. doi: 10.70099/cb/unah/2024.mem
ISBN del libro: 978-84-09-76685-7